La variante inglese.

Il rumore mediatico che sta causando l’isolamento in Inghilterra di una famiglia di varianti di SARS-CoV-2, propagata in molti nuovi casi di infezione e della quale si sospetta che possa trasmettersi più velocemente, merita un approfondimento, ora che qualche dato in più è disponibile dopo l’improvvido annuncio pubblico iniziale del ministro Hancock. Qui di seguito trovate una mia succinta trattazione dei dati disponibili (almeno di quelli che sono stato in grado di reperire), insieme a qualche mia considerazione. Il lettore tenga bene a mente che la situazione può evolvere molto velocemente, perchè la situazione non è affatto consolidata.
La ragione dell’allarme.
Qui di seguito vedete una slide che è stata mostrata quando i dati sono stati resi noti: rappresenta la percentuale della nuova variante reperita nei casi di nuova infezione, in cui si è usato il sequenziamento.
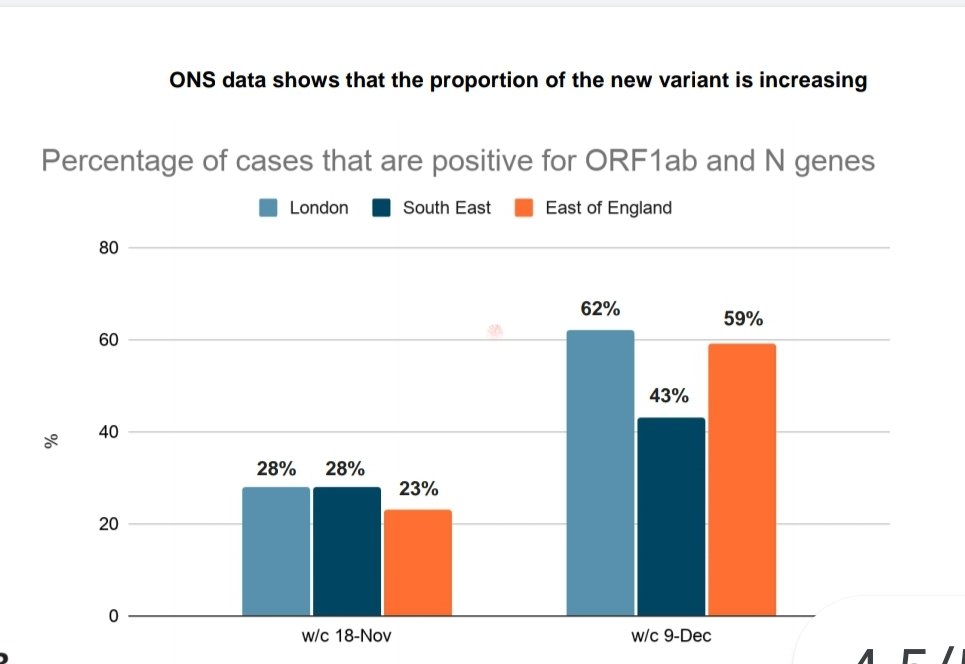
Come potete vedere, a Londra e nel Sud-Est dell’Inghilterra la famiglia di virus in questione è divenuta predominante.
Se guardiamo a Londra, nella settimana del 18 novembre si sono registrati 14.215 casi di infezione. Dalle percentuali riportate, otteniamo che 3.980 sono da imputarsi alla nuova famiglia di mutanti, mentre 10.235 sono infezioni dovute a tutti gli altri ceppi di SARS-CoV-2 circolanti a Londra nel periodo.
Nella settimana del 9 dicembre, sono stati registrati 41.496 casi; usando ancora le percentuali della slide, possiamo calcolare che 15.768 non sono appartenenti alla nuova famiglia, mentre 25.728 rappresentano i casi dovuti alle nuove varianti (un aumento di un fattore 6 rispetto al 18 novembre).
Un aumento della nuova famiglia pari al 9%, contro un aumento del resto dei ceppi virali pari al 2% (da cui deriva anche che Rt della nuova famiglia appare molto superiore alle precedenti varianti).
Una “nuova” famiglia di sequenze virali.
Sui giornali, si sta diffondendo rapidamente l’idea che in Inghilterra sia emerso un nuovo virus, con proprietà nuove.
Innanzitutto, non si tratta di un singolo virus, ma come avrete notato dal mio utilizzo di nomi collettivi, di una famiglia o di un “ramo” dell’albero filogenetico di SARS-CoV-2.
Questa famiglia è rappresentata nella figura seguente, ottenuta modificando la figura 1 di un lavoro del COVID-19 Genomics Consortium UK (CoG-UK) appena uscito.

Nella figura, ogni biforcazione dà origine a nuovi “rametti” di una porzione dell’albero evolutivo di SARS-CoV-2 ed ogni cerchietto rappresenta un particolare isolato virale, con le sue specifiche mutazioni. Il diametro dei cerchietti è proporzionale al numero di volte che quel definito virus è stato trovato, a partire da 1623 isolati virali sequenziati entro il 15 dicembre. Su sfondo grigio, si vede il ramo (in inglese “lineage”) della nuova famiglia o variante, che rispetto ai “parenti” più prossimi mostra 17 mutazioni aggiuntive. Come è agevole notare, il rettangolo grigio contiene una moltitudine di diversi cerchietti (174 per la precisione), i quali rappresentano ciascuno una specifica variante, che alle 17 mutazioni caratterizzanti la famiglia aggiunge qualche sua speciale variazione; ecco perchè non è corretto parlare di una singola variante, quanto di una nuova famiglia o di un nuovo ramo evolutivo di SARS-CoV-2. Per la precisione, questo è il lineage B.1.1.7.
E’ quindi scorretto definire B.1.1.7 come una “variante del virus”; ma è giusto indicarla come “inglese”? Neanche questo è appropriato. Nel riquadro bordato di rosso, che rappresenta un ingrandimento di una particolare porzione del lineage B.1.1.7, potete notare un cerchietto giallo. Quel colore indica che quella particolare sequenza, appartenente al lineage, è stata isolata al di fuori delle isole britanniche.
In particolare, virus che appartengono a questa famiglia sono stati identificati oltre che in Inghilterra anche in Danimarca, in Australia, in Sud Africa, in Olanda e probabilmente in alcune altre nazioni (come in Italia, dove di sicuro si è già identificata la sequenza di questa famiglia virale in un viaggiatore proveniente proprio dall’Inghilterra, e dunque potrebbe essere già arrivata anche prima). E’ vero che appartengono all’inghilterra la maggior parte degli isolati virali del lineage B.1.1.7 finora noti (tutti i pallini, eccetto quelli gialli, nella figura che abbiamo visto); ma questo dato è certamente influenzato dall’enorme numero di campioni sequenziati in Inghilterra rispetto alla maggioranza delle altre nazioni, visto che il solo consorzio COG-UK ha sequenziato oltre 140,000 isolati a partire da aprile in quel paese. In Inghilterra, il lineage B.1.1.7 è stato isolato per la prima volta su un campione del 20 settembre; ma in realtà non sappiamo se in altre parti del mondo virus di questo lineage non fossero già emersi, nè sappiamo se gli altri paesi siano stati contagiati con virus di tipo B.1.1.7 da viaggiatori inglesi o se non si accaduto invece il processo inverso.
Per questi motivi, ad oggi non è affatto scontato nè che B.1.1.7 sia un ramo evolutivo nuovo (anche se la sua emersione è certamente recente in Inghilterra), nè che la sua prevalenza sia maggiore in UK rispetto ad altrove, nè infine che sia originato in Inghilterra. Sono tutte inferenze ed ipotesi possibili, che possono però essere contrastate con altre possibilità egualmente probabili.
Perchè la discussione sulle origini di B.1.1.7 è importante? Perchè se questo ceppo stesse circolando già da tempo altrove nel mondo, oppure se – come risulta in Australia – si fosse estinto, allora sarebbe possibile ottenere molte maggiori informazioni sulle sue caratteristiche di trasmissibilità, virulenza e patogenicità, guardando retrospettivamente dati storici di molti paesi diversi; e, di fatto, significherebbe dire che non vi è qualcosa di realmente “nuovo” di cui preoccuparsi.
Come vedete, sto usando molti periodi ipotetici e sto facendo molte inferenze; questo, tuttavia, è il reale stato delle cose, e sbilanciarsi in un verso o nell’altro è prematuro.
Le mutazioni che caratterizzano B.1.1.7.
A questo punto, possiamo dare un’occhiata alle mutazioni condivise da tutti i membri del lineage B.1.1.7.
Come anticipato, questo gruppo è caratterizzato da 17 mutazioni principali: 14 mutazioni della sequenza delle proteine virali e tre delezioni. La tabella qui sotto le riassume tutte.
| gene | mutazione a livello genomico | mutazione a livello proteico |
|---|---|---|
| ORF1ab | C3267T | T1001I |
| C5388A | A1708D | |
| T6954C | I2230T | |
| 11288-11296 deletion | SGF 3675-3677 deletion | |
| spike | 21765-21770 deletion | HV 69-70 deletion |
| 21991-21993 deletion | Y144 deletion | |
| A23063T | N501Y | |
| C23271A | A570D | |
| C23604A | P681H | |
| C23709T | T716I | |
| T24506G | S982A | |
| G24914C | D1118H | |
| Orf8 | C27972T | Q27stop |
| G28048T | R52I | |
| A28111G | Y73C | |
| N | 28280 GAT->CTA | D3L |
| C28977T | S235F |
In tabella non sono indicate le mutazioni cosiddette “silenti”, cioè quelle che non producono conseguenze sulla struttura delle proteine virali; in maggioranza, queste sono quelle che differenziano i 174 appartenenti al ramo B.1.1.7 finora identificati.
La prima cosa da dire è che molte di queste mutazioni sono già note; è la loro combinazione in un gruppo così numeroso che non è stata osservata prima, e che rende il ramo B.1.1.7 ben distinto dal resto. Si osservi bene: ho deto “non osservato prima”, il che non vuol dire affatto che questo ramo non fosse presente in posti diversi dall’Inghilterra, ove l’imponente sforzo di sequenziamento ha portato alla sua identificazione e alla rapida definizione del suo progresso in termini numerici sul suolo inglese.
Nel dettaglio, possiamo osservare che ben 8 delle 17 mutazioni occorrono nella proteina spike (6 sostituzioni e due delezioni). Tra queste, si osserva la ben nota delezione 69-70 (ΔH69/ΔV70), di cui ho già discusso altrove, assurta all’onore delle cronache quando è stata ritrovata nei visoni danesi (pur se in realtà circolava anche altrove da tempo), ma importante soprattutto perchè conferisce resistenza a diversi anticorpi monoclonali. Nei visoni, questa delezione era emersa “accompagnata” da una serie di altre mutazioni nella proteina spike; e così è anche nel caso del ramo B.1.1.7, a suggerire un meccanismo di selezione comune, evidentemente guidato dalla selezione effettuata dal nostro sistema immunitario.
Vista la tendenza di questa mutazione a riemergere di continuo in posti diversi (è presente in oltre 3000 singoli genomi virali depositati nei database, in massima parte provenienti dall’Europa), spesso appunto in accompagnamento ad altre mutazioni nella regione RBD della proteina spike, è ovviamente ben diretto l’invito dei ricercatori inglesi a concentrarsi sulla sorveglianza dei virus che la portano.
Perchè la delezione 69-70 conferisce resistenza al riconoscimento da parte dei nostri anticorpi? Probabilmente, come è stato già messo in evidenza dai ricercatori del gruppo di Gupta in Inghilterra, perchè tale mutazione cade in una regione della proteina spike ben “visibile” al nostro sistema immune, trovandosi sulla superficie della proteina spike esposta al solvente (come si vede nella figura qui sotto, tratta da questo lavoro); il virus tende quindi a “perdere” quel frammento di proteina, così da offrire meno presa ai nostri anticorpi.

A parte questa delezione nella proteina spike, cosa altro sappiamo delle mutazioni del ramo B.1.1.7? Un buon riassunto si trova in questo lavoro appena uscito.
Innanzitutto abbiamo una mutazione già nota in posizione 501 della proteina spike, uno dei punti chiave di contatto fra il virus e il suo recettore nella zona RBD di spike. I dati sperimentali disponibili suggeriscono che la mutazione N501Y può aumentare l’affinità di spike per il recettore ACE2. Proprio questa mutazione, trovata nei topi di laboratorio usati per sviluppare alcuni vaccini, è risultata sensibile all’azione dei vaccini in sviluppo anche se associata anche ad un aumento dell’infettività e della virulenza.
E’ poi interessante la mutazione, sempre della proteina spike, P681H, che coinvolge uno dei 4 residui ricadenti nel sito di scissione della furina tra S1 e S2. Il sito di scissione della furina S1/S2 di SARS-CoV-2 non si trova nei coronavirus strettamente correlati ed è noto per promuovere l’ingresso nelle cellule epiteliali respiratorie e la trasmissione in modelli animali.
Le due mutazioni appena descritte (N501Y che P681H) sono state osservate indipendentemente, ma non per quanto ne sappiamo in combinazione.
Al di fuori della proteina spike, notiamo una mutazione (Q27stop) che elimina una delle proteine del virus, la proteina ORF8. All’inizio della pandemia, isolati di virus multipli con delezioni che portavano alla perdita di ORF8 sono stati isolati in tutto il mondo, incluso un grande cluster a Singapore. Il ceppo di Singapore, che aveva una delezione di 382 nucleotidi, era associato a un’infezione clinica più lieve e a una minore infiammazione post-infezione; tuttavia questo cluster si è estinto alla fine di marzo dopo che Singapore ha implementato con successo le misure di controllo. Ricerche successive hanno mostrato che la delezione di ORF8 ha solo un effetto molto modesto sulla replicazione del virus nelle cellule delle vie aeree primarie umane rispetto ai virus senza la delezione, portando a un leggero ritardo di replicazione rispetto ai virus con la delezione. Poiché ORF8 è solitamente lungo 121 amminoacidi, è probabile che il codone di stop alla posizione 27 osservato nella linea B.1.1.7 provochi la completa perdita di funzione della proteina.
Tiriamo le somme: domande e risposte.
Cosa possiamo concludere dalle informazioni di cui disponiamo?
Le misure eccezionali invocate in Inghilterra sono innanzitutto giustificate dall’aumento dei casi in quella nazione. In secondo luogo, di fornte all’isolamento del ceppo B.1.1.7, che appare in espansione in quel paese, è prudente non aspettare di sapere troppo al riguardo (cosa che richiede dati e presumibilmente anche danni), ma procedere subito con misure di sorveglianza stretta e di lockdown, intanto che aumentano le conoscenze a disposizione.
Mentre questo per gli scienziati è chiaro, non così per il pubblico; e quindi è bene ricordare che il fatto che B.1.1.7 appaia in espansione non significa poi molto, visto che tale espansione può giustificarsi non solo con un vantaggio genetico, ma anche con un fenomeno di “deriva” causato per esempio da un evento di disseminazione iniziale piuttosto ampio in una data località di un paese. Per intenderci: uno o più eventi di superdiffusione nel Kent di una variante specifica possono dare un temporaneo vantaggio a quella variante, senza che vi sia un sottostante reale vantaggio evolutivo (del tipo di una maggiore trasmissibilità). Questa è la ragione per cui il prof. Francois Balloux, per esempio, ci ricorda in un articolo pubblicato con il suo gruppo di ricerca su Nature che, di oltre 50.000 varianti analizzate, nessuna è finora risultata avere un vantaggio in termini di trasmissione, come potete qui.
Peraltro, sappiamo che apparentemente in Australia il ramo B.1.1.7 è comparso, ma è andato estinto (grazie alle misure di tracciamento e isolamento); e sappiamo che una delle 17 mutazioni che caratterizzano questo ramo, la mutazione Q27stop nella proteina Orf8, potrebbe portare all’estinzione il virus, come già accaduto per altre mutazioni simili sia in SARS che in una variante di SARS-CoV-2 apparsa a Singapore.
Quindi ecco la prima domanda e la prima risposta: il ramo evolutivo B.1.1.7 identificato in Inghilterra di recente ha un reale vantaggio in termini di trasmissibilità? Non lo sappiamo, abbiamo solo inferenze indirette in proposito.
Dal punto di vista della resistenza alla risposta anticorpale, e quindi ai vaccini, sappiamo che alcune delle 17 mutazioni del ramo B.1.1.7 conferiscono al virus una aumentata capacità di evadere la risposta immune (e dunque un vantaggio). Tuttavia, altre mutazioni comprese nella lista, quali quella in posizione 501 della proteina spike, sono già risultate ininfluenti ai fini del riconoscimento anticorpale, a dimostrazione che comunque i soggetti immunizzati sviluppano anticorpi in grado di riconoscere i mutanti che portano questa variazione. Abbiamo poi due altre evidenze indirette: finora, nella proteina spike sono state identificate oltre 4000 mutazioni, anche in combinazione multipla fra loro. Nessuna di queste, per quanto ne sappiamo, ha conferito resistenza ai vaccini in sviluppo. La seconda inferenza che possiamo fare riguarda il fatto che il ramo B.1.1.7 era già presente in Inghilterra mentre si ottenevano i dati per l’analisi ad interime dei risultati del trial 3 del vaccino Astra Zeneca poi pubblicati, ed era in quel paese in particolare in atto la sperimentazione con la “dose erronea” che poi ha portato ad una stima di protezione del 90% da parte del vaccino. I dati pubblicati sono stati ottenuti fino al 4 novembre; il ceppo B.1.1.7 circolava già dal 20 settembre; almeno nel periodo corrispondente, il nuovo ceppo non sembra aver avuto effetti significativi sul vaccino, anche se è vero che non aveva ancora raggiunto la prevalenza poi osservata a dicembre.
Infine, va ricordata la diversità dei vaccini in sviluppo, che coprono diverse zone della proteina spike, quando non ulteriori porzioni del virus (come nel caso dei vaccini proteici) o l’intero virus (nel caso di quelli ottenuti con virus attenuato o inattivato). Questa diversità aumenta (di molto) le probabilità che il virus non possa risultare in grado di evadere tutti i vaccini.
Ecco quindi la seconda domanda e la corrispodnente risposta: il ramo B.1.1.7 renderà inefficaci i vaccini in sviluppo? Non lo sappiamo se non in maniera indiretta ed inferenziale: le probabilità che questo avvenga ad oggi sembrano molto ridotte.
Veniamo ora alla patogenicità del ramo B.1.1.7. Nessuna delle mutazioni che contiene è nota per alterare in peggio la sintomatologia o il rischio di morte. Inoltre, a giudicare dai dati acquisiti in Inghilterra, non sembra che in contemporanea all’emersione del nuovo ramo evolutivo vi sia stato un peggioramento dei tassi di ospedalizzazione, di morte e di sintomatologia grave. Infatti, il capo della sanità pubblica inglese, prof. CHris Whitty, ha dichiarato quanto segue:
“There is no current evidence to suggest the new strain causes a higher mortality rate or that it affects vaccines and treatments although urgent work is underway to confirm this.”
Terza domanda, quindi, e terza risposta: il ramo B.1.17 è peggiore e più cattivo degli altri? Sembra di no (sembra, perchè questo è il risultato di una prima analisi dei dati disponibili).
Una raccomandazione finale.
Il virus SARS-CoV-2, come fanno i virus, muta di continuo. Ricordate la mutazione D614G, oggi divenuta predominante nel mondo? Oppure il gruppo di mutazioni emerse in Spagna, che questa estate hanno guadagnato terreno in Europa, e particolarmente in Inghilterra? Dobbiamo abituarci all’idea che, piuttosto frequentemente, isoleremo nuovi mutanti, perchè i virus funzionano così.
Cosa si fa quando si nota che alcuni di questi mutanti, come negli esempi citati, guadagnano terreno? Nell’attesa di capire qualcosa di più, si attuano misure di contenimento straordinario, allertando il resto del mondo. Anche questo, se ci pensate, è giusto.
Ciò che è profondamente sbagliato è soffiare sulle paure della gente, risuscitando ogni volta ipotesi possibili (in quanto ipotesi), ma del tutto indimostrate, soprattutto ipotesi negative fra le tante ipotesi possibili. E’ quello che ha fatto il ministro Hancock, nella sua prima comunicazione in cui non ha fornito lo straccio di un dato; è quello che hanno fatto i media italiani (tutti) rilanciando la notizia. Prima di avventurarsi nel descrivere ipotetiche resistenze a vaccini, virus cattivi e tutto il resto bisogna fermarsi a pesare i dati; e se i dati sono solo indiretti ed inferenziali, o se mancano, bisogna astenersi dal dipingere certi scenari.
Proprio come quando c’era chi si ostinava a dipingere virus che mutando sarebbero divenuti inevitabilmente più buoni, o a invocare l’omoplasia come meccanismo per un infallibile adattamento benigno, proprio allo stesso modo, con la stessa mancanza di dati, gli stessi salti logici e tutto sommato lo stesso modo di avanzare preconcetti oggi avviene per il perfido virus della perfida Albione (due ipotesi indimostrate in una frase).
Sereni, ma vigili: sequenziamo più che si può, isoliamo quanti più virus possibile, e facciamo scattare le opportune misure quando notiamo qualche virus che non conoscevamo prima in espansione; ma poi aspettiamo dati, ricerca e conferme, prima di fare i titoli dei giornali.


Un pensiero su “La variante inglese.”